Cassandra3 migration for Analytics AQS
Parent task.
| • Nuria | |
| Apr 8 2020, 6:41 PM |
| F34646232: image.png | |
| Sep 17 2021, 4:07 PM |
| F34646215: image.png | |
| Sep 17 2021, 3:54 PM |
| F34635767: image.png | |
| Sep 7 2021, 2:33 PM |
Cassandra3 migration for Analytics AQS
Parent task.
Script wmf-auto-reimage was launched by hnowlan on cumin1001.eqiad.wmnet for hosts:
['aqs1010.eqiad.wmnet']
The log can be found in /var/log/wmf-auto-reimage/202103091058_hnowlan_9533.log.
Completed auto-reimage of hosts:
['aqs1010.eqiad.wmnet']
Of which those FAILED:
['aqs1010.eqiad.wmnet']
Script wmf-auto-reimage was launched by hnowlan on cumin1001.eqiad.wmnet for hosts:
['aqs1010.eqiad.wmnet']
The log can be found in /var/log/wmf-auto-reimage/202103091139_hnowlan_16200.log.
Script wmf-auto-reimage was launched by hnowlan on cumin1001.eqiad.wmnet for hosts:
['aqs1011.eqiad.wmnet', 'aqs1012.eqiad.wmnet', 'aqs1013.eqiad.wmnet']
The log can be found in /var/log/wmf-auto-reimage/202103091243_hnowlan_10621.log.
Completed auto-reimage of hosts:
['aqs1012.eqiad.wmnet']
Of which those FAILED:
['aqs1012.eqiad.wmnet']
Script wmf-auto-reimage was launched by hnowlan on cumin1001.eqiad.wmnet for hosts:
['aqs1012.eqiad.wmnet', 'aqs1014.eqiad.wmnet', 'aqs1015.eqiad.wmnet']
The log can be found in /var/log/wmf-auto-reimage/202103091411_hnowlan_3565.log.
Completed auto-reimage of hosts:
['aqs1012.eqiad.wmnet', 'aqs1014.eqiad.wmnet', 'aqs1015.eqiad.wmnet']
and were ALL successful.
Change 675174 had a related patch set uploaded (by Hnowlan; author: Hnowlan):
[labs/private@master] profile::aqs_next: add stub password
Change 675174 merged by Hnowlan:
[labs/private@master] profile::aqs_next: add stub password
Script wmf-auto-reimage was launched by hnowlan on cumin1001.eqiad.wmnet for hosts:
['aqs1012.eqiad.wmnet']
The log can be found in /var/log/wmf-auto-reimage/202105180958_hnowlan_17932.log.
Change 719233 had a related patch set uploaded (by Btullis; author: Btullis):
[operations/puppet@production] Add a promehtheus scrape target for the aqs_new role
Change 719233 merged by Btullis:
[operations/puppet@production] Add a prometheus scrape target for the aqs_next role
Here is the migration plan document: https://docs.google.com/document/d/1FGub_rRIrv77Miadp0Muvf6EwpbvcW2dtZ_qICSt-2o/edit
The snapshot is copying to the new hosts now. We'll be able to keep an eye on progress by monitoring the usage of the /srv/cassandra-a and /srv/cassandra-b filesystems on these two graphs.
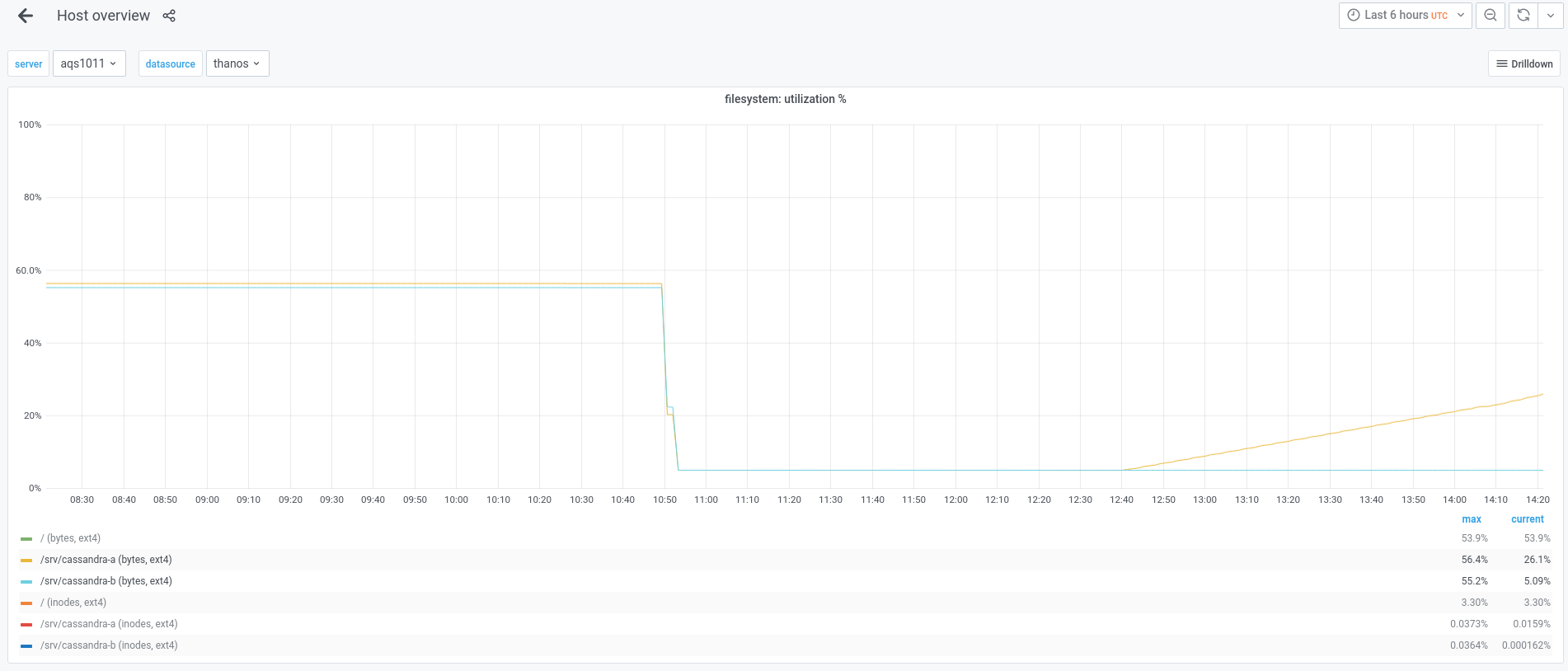
Once the copy operation is complete, we will want to perform a validation of the contents of the directories, before restoring the snapshots.
The transfer operation of the snapshots completed successfully.
I have done some additional verification to make sure that the snapshots are consistent.
find /srv/cassandra-b/data/local_*/*/snapshots/1631011478226 -type f > aqs1004-cassandra-b-1631011478226-files.txt`
time nice sha1sum `shuf -n 1000 aqs1004-cassandra-b-1631011478226-files.txt` > aqs1004-cassandra-b-1631011478226-1000-random-files.chk
cat aqs1004-cassandra-b-1631011478226-files.txt|sed 's/\//@/8; s/\//_/g; s/@/\//; s/1631011478226\//1631011478226\/1631011478226\//' > aqs1004-cassandra-b-1631011478226-files_modified.txt cat aqs1004-cassandra-b-1631011478226-1000-random-files.chk|sed 's/\//@/8; s/\//_/g; s/@/\//; s/1631011478226\//1631011478226\/1631011478226\//' > aqs1004-cassandra-b-1631011478226-1000-random-files_modified.chk
for i in $(cat aqs1004-cassandra-b-1631011478226-files_modified.txt); do if ! [ -f $i ]; then echo $i does not exist; fi ; done
sha1sum --quiet -c aqs1004-cassandra-b-1631011478226-1000-random-files_modified.chk
Given that these tests pass for all four of the snapshots, plus we understand that transfer.py also carried out checksumming, I'm happy that these are consistent and we can press on with the importing of keyspaces at any time.
The one thing that we have to keep an eye on is the capacity of the drives, given that a snapshot takes up ~60% of each of the two available file systems. Loading a full snapshot might take us up to 90% by my reckoning, so we may need to be careful about this.
Change 721849 had a related patch set uploaded (by Btullis; author: Btullis):
[operations/puppet@production] Add temporary rsync modules to two Cassandra nodes
We have decided to use rsync for the next transfer from the v2 cluster to the v3 cluster.
As such I'm proposing to create temporary rsync modules on the two destination hosts: https://gerrit.wikimedia.org/r/c/operations/puppet/+/721849
Mentioned in SAL (#wikimedia-analytics) [2021-09-17T15:15:01Z] <btullis> btullis@aqs1004:~$ sudo nodetool-a repair --full && sudo nodetool-b repair --full (T249755)
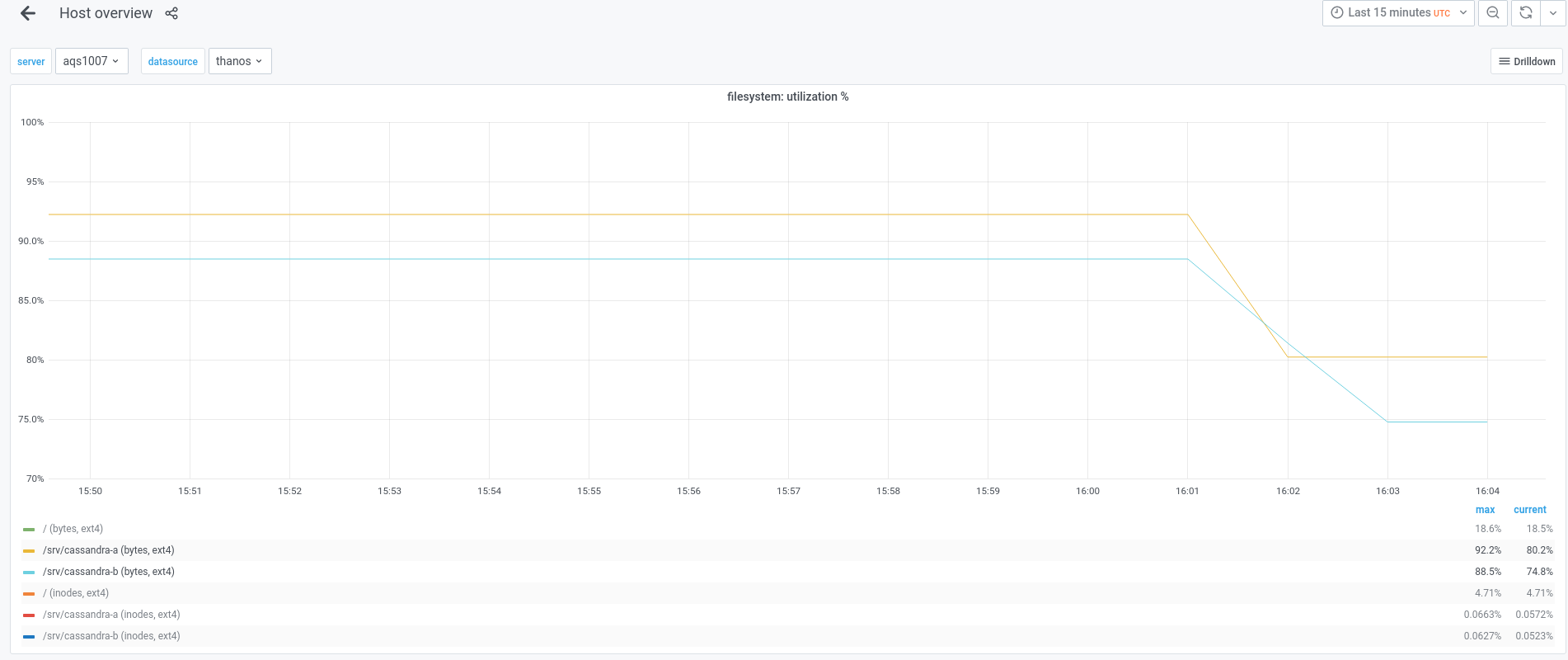
I think that we might need to remove those previously created snapshots, because the usage on aqs1004 and aqs1007 is above 92%.
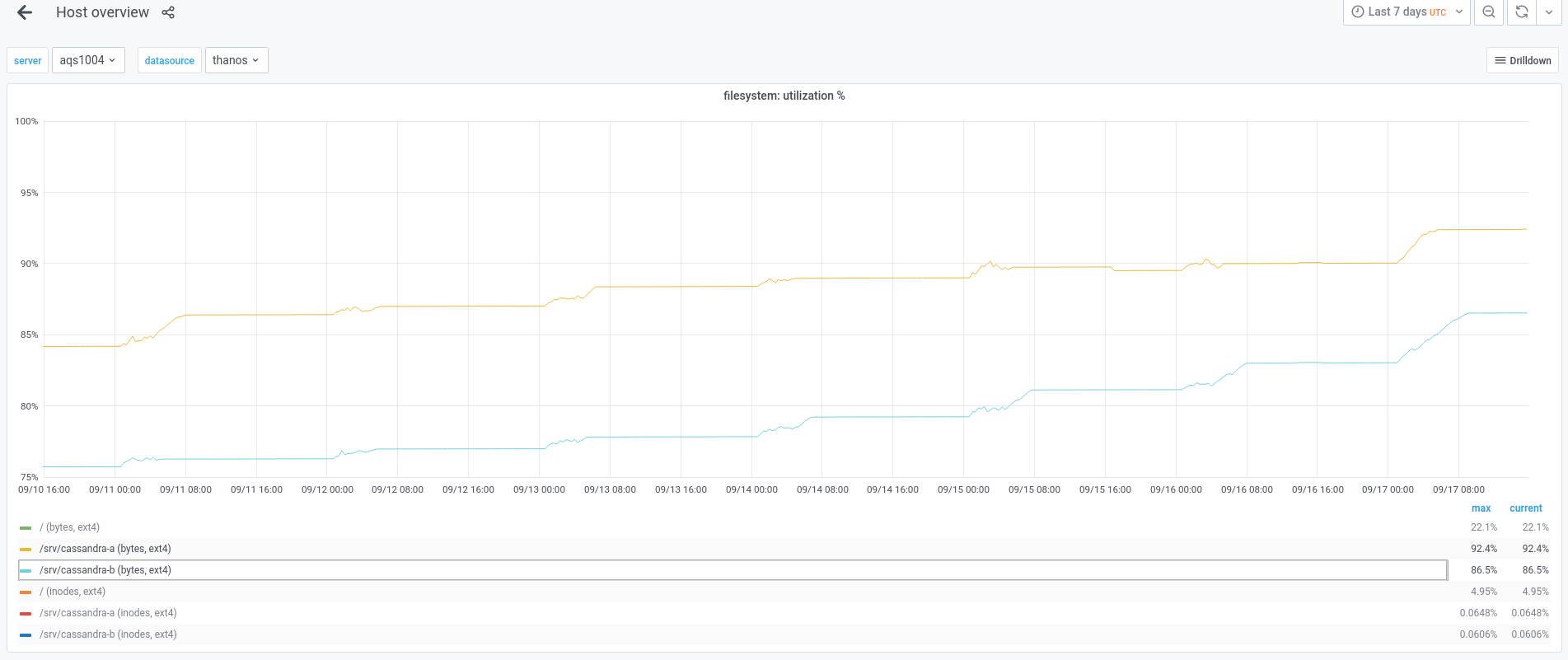
Mentioned in SAL (#wikimedia-analytics) [2021-09-17T16:03:00Z] <btullis> Cleared all snapshots on aqs100[47] to reclaim space with nodetool-[ab] clearsnapshot (T249755)
Change 721849 merged by Btullis:
[operations/puppet@production] Add temporary rsync modules to two Cassandra nodes